In several of my projects, I have to focus on specific regions of a genome, let’s say while looking for binding peaks. It is quite common to download a full genome as a fasta file, or each chromosome separately from Ensembl or NCBI. However, it is difficult to get a specific region of a genome (chromosome, start, end). I didn’t find any easy solution for it so far. Therefore, I focused on using tools available directly from NCBI, the e-utilities.
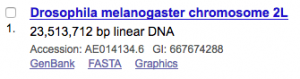
To get a specific region of the genome one needs to know the GI identifier of the chromosome of interest. Let’s say we are interested in sequences from chromosome 2L of Drosophila melanogaster. The first step is to search for it on the Nucleotide database of NCBI.
![]()
Here, we have to note the GI identifier (667674288)
Then, the command to run is
efetch -db=nuccore -format=fasta -id=667674288 -seq_start=12200 -seq_stop=13000
This will return the sequence of chromosome 2L between positions 12200 and 13000 in fasta format as follow:
>AE014134.6:12000-12200 Drosophila melanogaster chromosome 2L
ATTTCATATGTATTCACAACTTCATCACATGCGTAGGTTTCCTTATTATTCTCTGCATCATTTTCTTCCA
ACACAGACTCTTCATTTTCTAATGGTTCTACTGGAACAACGCCAGTGGGTTGCACGACTCTGGACGTGGC
TAAAGCAATACGCTGCAGTTCAGAAGAAGACATCATATACAGTGCTTCTCCAGAGTTTGTA
This command gets really handy when working with multiple regions. One could save all the regions of interest in a tab separated file with one column containing the start position and the other the end positions such as:
12000 12200
13500 13700
…
and then use awk to get the sequences for all those regions and save it as a fasta file:
awk '{ system("efetch -db=nuccore -format=fasta -id=667674288 -seq_start="$1" -seq_stop="$2" ") }' dmel_chr2L_peaks.txt > dmel_chr2L_peaks.fasta
Enjoy and let me know if you have any alternative solution for this problem.